

Acute myeloid leukemia (AML) is an aggressive heterogeneous hematologic disease with high mortality in patients older than 60 years. Clinical studies have proven that combinations of FDA-approved Bcl-2 inhibitor, venetoclax and hypomethylating agents (Ven/HMA) are highly effective in elderly patients with AML (DiNardo et al., 2019). Despite improved remission rates, the duration of response is still inadequate. Adhesion to the bone marrow (BM) niche is critical for AML initiation, progression and leukemic stem cell (LSC) survival after induction therapy. The vascular adhesion molecule, E-selectin (E-sel) is responsible for the tethering and rolling of leukocytes on perivascular endothelial BM niche cells (EC). In leukemia, E-sel has crucial roles in BM homing and engraftment (Krause et al, 2006).
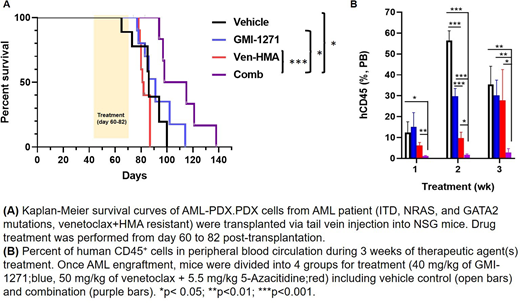
Through patient-derived AML xenograft (PDX) models and single cell proteomics, we have elucidated the roles of E-sel in AML survival and drug resistance. A PDX model derived from a patient who had developed resistance to Ven/HMA were treated with the E-sel antagonist, GMI-1271 (uproleselan; GlycoMimetic, Inc). We found that targeting E-sel mobilized human AML cells and sensitized them to Ven/HMA. The number of circulating leukemic cells was significantly reduced by combinatorial treatment of GMI-1271 with Ven/HMA in comparison to Ven/HMA alone (p < 0.05). The synergistic effects of the combinatorial treatment on AML-PDX mouse survival were determined by Kaplan-Meier analysis. The combination of GMI-1271 and Ven/HMA significantly prolonged the survival of mice compared to vehicle control (p = 0.015) as well as the Ven/HMA (p = 0.0009) and GMI-1271 groups (p = 0.03). The median survival of the vehicle control, GMI-1271, Ven/HMA, and combination-treated groups of mice was 86, 91, 81.5, and 106.5 days, respectively. Histological analysis of BM, spleen, lung and liver demonstrated differences in leukemia cell infiltration, confirming enhanced anti-leukemia efficacy of the combination treatment.
To delineate the mechanism of E-sel at the onset of drug mediated changes in AML signaling signatures, we employed another PDX model (Flt3-ITD and WT1 mutations, sorafenib-resistant). PDX mice with advanced AML (more than 20% human AML cells circulation in peripheral blood) were administered Ven/HMA, GMI-1271, or combination for 2 days. Single cell proteomics analysis by CyTOF determined that combinatorial treatment diminished levels of Ki67, IDU, and pRb compared to vehicle control or Ven/HMA alone, resulting in decreased proliferation of AML blasts. Activation of eNOS to produce nitric oxide (NO) through PI3K/AKT kinase, maintains clonogenic cell growth in malignant cells. A recent publication has demonstrated that introduction of NOS blockers in combination with chemotherapy led to slower leukemia progression and longer remissions in contrast to chemotherapy alone (Passaro et al, 2017). Interestingly, we observed reduced activation of PI3K and AKT in AML blasts as well as in BM CD31+EC cells in the GMI-1271 treated PDX model. eNOS phosphorylation was subsequently decreased in EC, suggesting that inhibition of E-sel may protect BM vasculature by blocking the production of NO. In addition, targeting E-sel showed signaling alterations in AML MSC. Administration of E-sel antagonist increased mTOR expression in MSC from AML-PDX. Combination treatment induced higher Ki67 positivity, and hyperactivation of pRb and p-S6 in MSC in vivo. Our group and others have recently reported that Ven-resistant AML cells exhibit an increased dependence on alternate anti-apoptotic proteins, Mcl-1 and Bcl-xl (Konopleva et al., 2016). We found that concomitant treatment in vivo with GMI-1271 and Ven/HMA further decreased the expression of Bcl-xl and Mcl-1 in AML blasts compared to Ven/HMA alone, suggesting a critical role for E-sel antagonists in overcoming drug resistance. E-selectin binding potential and focal adhesion kinase activity in AML blasts were decreased upon acute administration of pharmacological E-sel inhibitor. Other oncogenic signaling pathways including MAPK, p-S6, and STAT3, were all inhibited by the addition of GMI-1271 to Ven/HMA. Collectively, our results provide first evidence that an E-sel targeting strategy with GMI-1271 can overcome microenvironmental resistance to Ven/HMA-based therapy in AML by cancer cell autonomous and non-cell autonomous mechanisms in the BM vascular niche.
Fogler:GlycoMimetics: Current Employment, Current equity holder in publicly-traded company, Patents & Royalties. Magnani:GlycoMimetics, Inc.: Current Employment, Current equity holder in publicly-traded company, Membership on an entity's Board of Directors or advisory committees, Patents & Royalties. Andreeff:Daiichi-Sankyo; Breast Cancer Research Foundation; CPRIT; NIH/NCI; Amgen; AstraZeneca: Research Funding; Centre for Drug Research & Development; Cancer UK; NCI-CTEP; German Research Council; Leukemia Lymphoma Foundation (LLS); NCI-RDCRN (Rare Disease Clin Network); CLL Founcdation; BioLineRx; SentiBio; Aptose Biosciences, Inc: Membership on an entity's Board of Directors or advisory committees; Amgen: Research Funding; Daiichi-Sankyo; Jazz Pharmaceuticals; Celgene; Amgen; AstraZeneca; 6 Dimensions Capital: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal